Welcome to learnMSA’s documentation!
Our tool is under active development and feedback is very much appreciated.
Installation
You have 3 options to install learnMSA.
Option 1: Singularity/Docker
We provide a hassle-free Docker image including everything you need to align on GPU with protein language model support.
This is the recommended and most stable way to install learnMSA.
singularity build learnmsa.sif docker://felbecker/learnmsa
singularity run --nv learnmsa.sif learnMSA
Running the container with --nv is required for GPU support.
Option 2: Conda/mamba and pip
Create a conda environment:
conda create -n learnMSA python=3.12
conda activate learnMSA
Install learnMSA (CUDA toolkit included):
pip install learnMSA
Additional installs for sequence weights (recommended!):
conda install -c conda-forge -c bioconda "python=3.12.*" mmseqs2
You may have to set up Bioconda channels. We pin the currently installed python executable to prevent installing newer mmseqs2 versions that would remove or alter.
Additional installs for language model support (recommended!):
pip install torch==2.6
(optional) Verify that TensorFlow and learnMSA are correctly installed:
python3 -c "import tensorflow as tf; print(tf.__version__, tf.config.list_physical_devices('GPU'))"
learnMSA -h
Option 3: Bioconda
conda create -c bioconda -n learnMSA learnMSA
This installs everything you need in a conda environment. However, due to the way TensorFlow is distributed via conda currently no GPU support is provided out of the box.
Therefore, a post install fix is needed:
conda activate learnMSA && pip install tensorflow[and-cuda]=="$(pip show tensorflow | grep ^Version: | awk '{print $2}')"
Using learnMSA
Getting started
Get an overview of the command line options with:
learnMSA -h
Get help for a specific option with:
learnMSA help [OPTION]
MSA
Recommended way to align proteins with learnMSA version >= 2.0.10:
learnMSA -i INPUT_FILE -o OUTPUT_FILE –use_language_model
Recommended way to align proteins with learnMSA version < 2.0.10:
learnMSA -i INPUT_FILE -o OUTPUT_FILE –use_language_model –sequence_weights
Without language model support (recommended for speed and for proteins with very high sequence similarity):
learnMSA -i INPUT_FILE -o OUTPUT_FILE
Note: If you installed learnMSA via docker/singularity, you have to run singularity run --nv learnmsa.sif learnMSA -i ...
Note 2: Since v2.0.10 the default behavior changed: Sequence weights are now used by default and the --sequence_weights option was removed. Instead, a --no_sequence_weights option exists to align without sequence weights (not recommended). Users that installed learnMSA via pip have to manually install mmseqs2 (conda is recommended, see above).
To output a pdf with a sequence logo alongside the msa, use --logo. For a fun gif that visualizes the training process, you can use --logo_gif (attention, slows down training and should not be used for real alignments).
Interactive notebook with visualization:
Run the notebooks learnMSA_demo.ipynb or learnMSA_with_language_model_demo.ipynb with juypter.
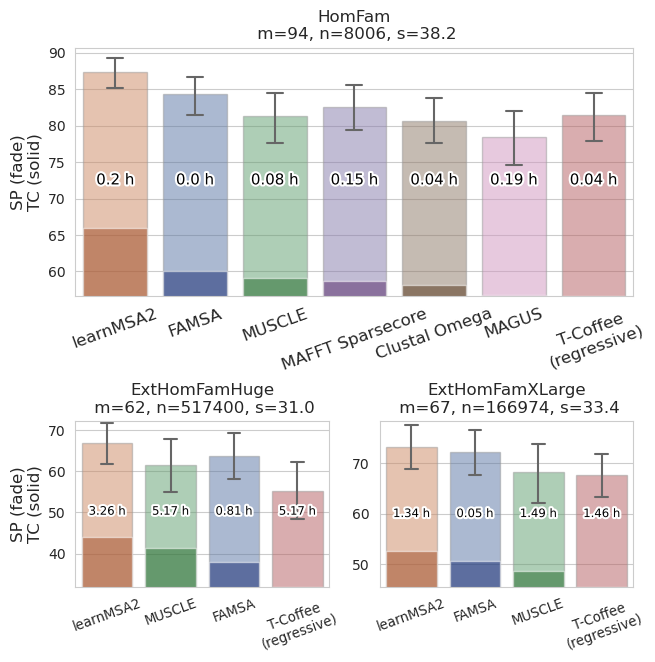
Benchmark:
Publications
Becker F, Stanke M. learnMSA2: deep protein multiple alignments with large language and hidden Markov models. Bioinformatics. 2024
Becker F, Stanke M. learnMSA: learning and aligning large protein families. GigaScience. 2022
${\text{\color{red}Troubleshooting:}}$
Error:
tensorflow.python.framework.errors_impl.UnknownError: {{function_node __wrapped__Expm1_device_/job:localhost/replica:0/task:0/device:GPU:0}} JIT compilation failed.
Your root error is:
TensorFlow libdevice not found
Fix:
Find nvvm directory:
find / -type d -name nvvm 2>/dev/null
Expected outputs:
<path>/nvvm
If there are multiple paths, choose the one matching your conda environment.
Run:
export XLA_FLAGS=--xla_gpu_cuda_data_dir=<path>
Error:
ERROR: Flag 'minloglevel' was defined more than once (...)
Fix:
pip install --no-deps --upgrade sentencepiece==0.1.99
User Guide:
API Reference: